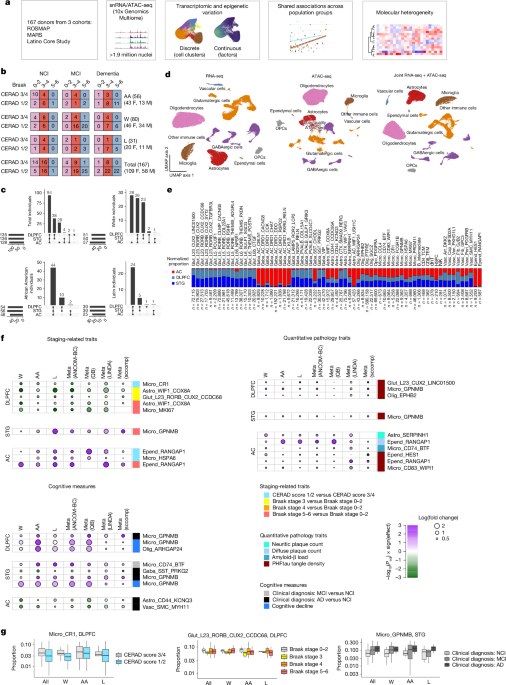
Choroba Alzheimera (AD) pozostaje najczęstszą przyczyną demencji u osób starszych, a jej złożone podłoże prowadzi do postępującej neurodegeneracji i utraty pamięci. Choć badania pośmiertne tkanek mózgowych pozwoliły zidentyfikować główne patologie, takie jak złogi amyloidu-beta oraz splątki białka tau, większość dotychczasowych analiz genomowych skupiała się na populacjach pochodzenia europejskiego. W obliczu różnic w zapadalności na tę chorobę między grupami etnicznymi, kluczowe stało się ustalenie, czy dotychczasowe odkrycia dotyczące zmian w poszczególnych typach komórek mózgowych są uniwersalne.
Najnowsze badanie, opublikowane w lipcu 2026 roku, wypełnia tę lukę, analizując próbki tkanek mózgowych od 167 osób, które identyfikowały się jako Afroamerykanie, Latynosi lub osoby rasy białej. Naukowcy wykorzystali zaawansowane techniki sekwencjonowania RNA pojedynczych jąder (snRNA-seq) oraz analizy dostępności chromatyny (snATAC-seq), aby zbadać trzy regiony mózgu: korę przedczołową grzbietowo-boczną, górny zakręt skroniowy oraz przednią część jądra ogoniastego.
Wspólne sygnatury komórkowe w chorobie Alzheimera
Badanie wykazało, że pewne zmiany w składzie komórkowym mózgu są wspólne dla wszystkich badanych grup populacyjnych, niezależnie od pochodzenia. Najsilniejszym sygnałem była zwiększona obecność specyficznej podgrupy mikrogleju (GPNMB+) w korze mózgowej u osób z kliniczną diagnozą demencji oraz szybszym tempem pogorszenia funkcji poznawczych. Mikroglej ten wykazuje wysoką ekspresję genów związanych z metabolizmem lipidów, migracją komórek i reorganizacją naczyń krwionośnych.
Wśród innych istotnych zmian zaobserwowanych we wszystkich grupach można wymienić:
Znaczenie analizy ciągłej ekspresji genów
Tradycyjne podejście, polegające na przypisywaniu jąder komórkowych do sztywnych klastrów, może pomijać subtelne, ale istotne zmiany w funkcjonowaniu komórek. Dlatego autorzy zastosowali analizę czynnikową, która pozwoliła zidentyfikować ciągłe wzorce ekspresji genów, szczególnie widoczne w astrocytach i oligodendrocytach.
Okazało się, że niektóre czynniki (np. czynnik 4 w oligodendrocytach) są silnie powiązane z nasileniem patologii, takich jak liczba płytek neurytycznych czy gęstość splątków białka tau. Co istotne, niższa ekspresja czynnika 6 w astrocytach, odpowiedzialnego za transport glutaminianu, sugeruje, że w chorobie Alzheimera dochodzi do poważnych zaburzeń na poziomie synaptycznym.
Molekularna heterogeniczność pacjentów
Jednym z najważniejszych wniosków badania jest identyfikacja sześciu odrębnych podgrup molekularnych wśród osób z zaburzeniami poznawczymi. Podgrupy te nie pokrywają się w pełni z tradycyjną klasyfikacją neuropatologiczną, co oznacza, że pacjenci z podobnym stopniem zaawansowania zmian w mózgu mogą wykazywać odmienne profile molekularne.
Odkrycie to sugeruje, że zbieżne objawy kliniczne u różnych pacjentów mogą wynikać z rozregulowania odmiennych ścieżek molekularnych. Może to mieć kluczowe znaczenie dla przyszłych terapii celowanych oraz precyzyjnej stratyfikacji pacjentów w badaniach klinicznych.
Ograniczenia i perspektywy
Autorzy podkreślają, że badanie ma charakter obserwacyjny, a uzyskane wyniki stanowią punkt wyjścia do dalszych prac. Mimo że liczba przeanalizowanych próbek (399) jest znacząca, ograniczenia w głębokości sekwencjonowania snATAC-seq utrudniły identyfikację specyficznych dla populacji różnic w dostępności chromatyny. Jedynym wyjątkiem był locus w pobliżu genu KANSL1, gdzie zaobserwowano różnice w dostępności chromatyny między Afroamerykanami a osobami rasy białej, co jednak nie przekładało się bezpośrednio na zmiany w ekspresji tego genu.
Badanie to stanowi istotny krok w kierunku zrozumienia biologicznych podstaw choroby Alzheimera w zróżnicowanych populacjach. Wskazuje ono, że choć istnieją wspólne mechanizmy neurodegeneracji, to różnorodność molekularna pacjentów wymaga uwzględnienia w procesie opracowywania nowych metod diagnostycznych i terapeutycznych.

{kind=link}